
本文由CIAPH根据路璐女士在2018CIAPH医药研发数字化北京站会议演讲整理
2018年11月16日,CIAPH医药研发数字化北京站会议顺利召开,会议由来自医疗研发、药品研发、CRO、医药独立顾问等多位演讲人,分享各自领域的精彩实践。医药研发领域独立顾问路璐女士,就eCTD电子申报系统做了全面讲解,话题涉及政策监管背景、eCTD介绍、技术要求以及研发药企IT应该做好的准备,受到参会者广泛关注,本文根据路璐女士现场演讲整理而成。

医药研发领域独立顾问 路璐
一、什么是eCTD
1、什么是CTD
医药研发的周期很长,涉及到的系统应用很多,eCTD是其中很重要的一项,可以说,eCTD标志着一个全新的时代到来,因此,药企研发相关的同仁,都需要熟悉和运用eCTD,而作为企业系统搭建者的IT,更要为新时代的到来做好准备。
众所周知,eCTD由CTD发展而来。CTD(Common Technical Document)是国际公认的文件编写格式,用来制作一个向药品注册机构递交的结构完善的注册申请文件,是由ICH为了解决人用药在申请注册中格式和内容的统一,而决定采用的统一的注册申请文件格式。
ICH(International Conference of Harmonization)是由美国,欧洲和日本三方发起的国际协调会议组织,其成立的背景是:以上三方发起主体,在各自区域内对于在人用药申请注册的技术要求方面,已经取得了相当大的协调统一,但直到目前为止,各国对于注册申请文件,仍然没有一个统一的格式,每个国家对于提交的技术报告的组织及文件中总结和表格的制作,都有自己的要求。为解决这些问题,ICH决定采用统一的格式来规范各个地区的注册申请,并在2003年7月起首先在欧洲实行,这就是常规技术文件CTD。ICH成立的宗旨,是以统一的标准格式,减少药企在研发方面的重复工作及资金投入。
2、ICH的成员
路总指出,“ICH本质上是医药研发前瞻性的国家及区域,发起的协调组织,制定和执行统一的注册申请标准,同时,要求加入的成员统一执行,以避免格式差异、内容差异和研发工作的重复”。
ICH委员会下面,有很多主体成员,国家及行业协会都可以作为成员:
创始监管机构成员(Founding Regulatory Members):欧盟委员会(EC)、日本厚生劳动省/日本药品及医疗器械管理局(MHLW/PMDA)及美国食品药品管理局(FDA);
创始行业成员(Founding Industry Members):欧洲制药工业协会联合会(EFPIA)、日本制药工业协会(JPMA)、美国药品研究与制造商协会(PhRMA);
常任监管机构成员(Standing Regulatory Members):加拿大卫生部(Health Canada)、瑞士医药管理局(Swissmedic);
监管机构成员(Regulatory Members):巴西卫生监督局(ANVISA)、中国国家食品药品监督管理总局(CFDA)、新加坡卫生科学局(HSA)、韩国食品药品安全部(MFDS)、台湾卫生福利部食品药物管理署(TFDA);
这里要指出的是,中国国家食品药品监督管理总局(CFDA),是于2017年6月,加入ICH的,2018年6月,成功当选为ICH管理委员会成员的。
常任观察员(Standing Observers):国际制药企业和协会联合会(IFPMA)、世界卫生组织(WHO);
立法或行政机构(Legislative or Administrative Authorities):印度中央药品标准控制组织(CDSCO)、古巴国家药品和医疗器械控制中心(CECMED)、墨西哥联邦卫生风险保护局(COFEPRIS)、哥伦比亚国家药品和食品监督局(INVIMA)、摩尔多瓦药监局(MMDA)、哈萨克斯坦国家中心(National Center)、马来西亚国家药品管理局(NPRA)、俄罗斯联邦卫生监督局(Roszdravnadzor)、南非健康产品管理局(SAHPRA)、亚美尼亚药物和医疗技术专业科学中心(SCDMTE)、澳大利亚治疗产品管理局(TGA)、土耳其药品和医疗器械机构(TITCK)。
3、ICH的任务和目标
作为欧盟、美国、日本三方发起的协调组织,ICH在演进的过程中,不断完善成为功能健全且成员众多的组织,其任务和目标也很清晰:
为在药品注册与注册维护的技术指导原则和要求的解释与应用中为实现更大程度的协调提出建议;
论坛的维护,就药品技术要求的协调在监管机构和医药行业间构建科学问题对话;
出于国际视角,为保护公众健康作出贡献;
监测与更新协调的技术要求,使研发数据更大程度的相互接受;
因治疗的进步和药品生产新技术的发展,通过对需要议题的协调,避免未来要求的不同;
促进采纳新的或改善的技术研究与开发方法,更新或替代目前作法;
通过协调的指导原则及其使用方面信息的传播、交流及培训,鼓励通用标准的实施与接轨;
制定ICH监管活动医学词典(MedDRA)术语的政策,同时确保MedDRA作为方便人用药品国际监管信息分享的标准化词典的科学与技术维护、开发和传播。
4、ICH指导原则、制修订工作程序
在ICH里面,要制定某一项规范,就要经过以下工作程序:
第1阶段构筑共识:新议题提案经大会批准成为新议题后,成立专家工作组。专家工作组依据概念文件和业务计划,不断讨论形成第1阶段技术文件(Step 1 Technical Document),即指导原则草案的基础。
第2阶段:
第2a阶段确认共识:第1阶段的技术文件经大会批准后,全体成员、大会成员将会对技术文件共识进行确认,形成第2a阶段终版技术文件(Step 2a Final Technical Document)。
第2b阶段采纳指导原则草案:根据第2a阶段终版技术文件,制定第2b阶段指导原则草案(Step 2b Draft Guideline),并由ICH大会监管机构成员确认的过程。
第3阶段监管机构征求意见和讨论:ICH各地区/国家的监管机构对指导原则草案公开征求意见。根据反馈的意见,专家工作组进行讨论,修改指导原则草案。
第4阶段采纳指导原则:大会监管机构成员对指导原则草案达成最终一致,并通过。
第5阶段实施指导原则:ICH各地区/国家监管机构通过各自行政程序实施指导原则。
5、eCTD当前的执行情况
每个国家和地区执行eCTD的时间和进度是不一样的。下表是一个关于不同国家执行eCTD的情况统计表:
接受eCTD是指该国家接受eCTD格式的时间,而强制执行是指必须采用统一格式文件进行申请注册。在接受和强制执行中间,有一个比较漫长的周期,在这个周期内,申请注册的国家,可以采用eCTD也可以采用纸质文件申请注册。强制执行时间后,则必须是采用eCTD的格式。通过下图可以看出,两者之间的周期越来越快,可见eCTD代表的新时代即将来临。
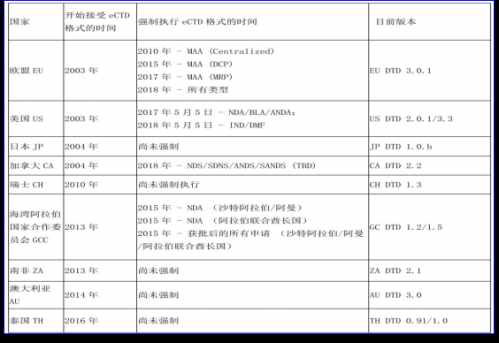
当前,正在建设和筹备eCTD的国家有:China中国,Egypt埃及,Jordan约旦,Tanzania坦桑尼亚,Israel以色列。正在探讨计划eCTD的有:Brazil巴西, India印度,Russia俄罗斯,Ukraine乌克兰,Mexico墨西哥,Singapore新加坡,South Korea韩国等。
6、eCTD作用的阶段
在新药研发的过程中,从实验室研究筛选出活性分子或者化合物,进入到临床前研究的毒理、药理等实验,下一步即是临床申请进入临床试验阶段,从此开始产生的各种数据,包括Ⅰ期、Ⅱ期、Ⅲ期临床实验,直到上市许可申请获批前,将会产生各种各样巨量的数据。这些数据都是申请过程中,不可或缺的数据。
这些数据,在申请上市许可时候,需要按照法规要求提交给药品监管机构,如FDA、CDE(国家食品药品监督管理总局药品评审中心)等。eCTD是将新药研发各个阶段中,从源头数据开始到上市申请许可中间的数据,整合成文件提交给药品监管部门。
因为医药研发的前期投入成本已经非常高昂,经过的试验阶段与时间周期非常漫长,而报批的准确性需要非常精准,因此,在越来越严格的监管背景下,eCTD在医药研发中的重要性,日益凸显。同时,报批阶段的时效性与经济获益是绝对成正比的,eCTD对于新药研发企业的价值,是必须引起重视的。
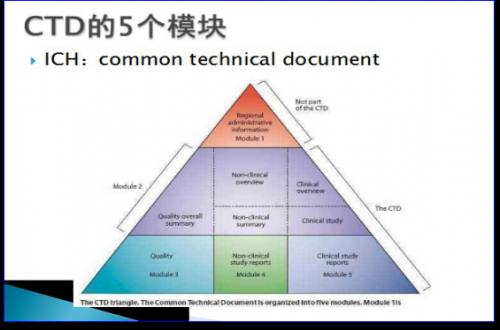
7、CTD的5个模块
CTD文件(Common Technical Document)是国际公认的文件编写格式,用来制作一个向药品注册机构递交的结构完善的注册申请文件,共由五个模块组成,模块1是地区特异性的,模块2、3、4和5在各个地区是统一的。
模块1,行政信息和法规信息:包括对各地区特殊的文件,例如申请表或在各地区被建议使用的标签,其内容和格式可以由每个地区的相关注册机构来指定。
模块2,CTD文件概述:2.1CTD总目录;2.2申请药品的一般介绍(药理作用、临床适应症);2.3-2.7质量概要、临床前综述、临床综述、临床前研究列表总结和表格概要、临床研究概述。
模块3,质量部分:文件提供药物在化学、制剂和生物学方面的内容。
模块4,非临床研究报告:文件提供原料药和制剂在毒理学和药理学试验方面的内容。
模块5,临床研究报告:文件提供制剂在临床试验方面的内容。
8、药品注册文件递交的四种方式
随着医药研发的演进,注册文件递交的方式,总共经历了四种变化:
纸质:采用快递形式,运送大量的纸质文件。
PDF文件:做成PDF文件后,用光盘形式提交。其缺陷是数据是非结构化的,想要建立关联或者进行搜索时,非常困难。
NeES:光盘的形式,在数据结构方面比PDF有了提升。
eCTD:包含大量超链接、文件格式等,可以帮助做一些格式化的审核。
9、监管的痛点
监管机构的评审效率,也关乎新药获批的进程。监管机构在评审时候遇到的痛点,包括以下几个方面:提交数量日渐增多;数据质量缺乏规范;统计和监测困难;数据量巨大;数据安全性和透明度要求增加。
二、eCTD的技术要求
1、国家监管机构对eCTD 格式注册申报的要求
国家监管机构对eCTD 格式注册申报的要求,包括以下三个方面: eCTD 格式注册申报的信息可以在监管体系中被处理,没有错误的信号信息;药品研究单位可以提供eCTD 格式注册申报提交的资料目录;eCTD 格式注册申报信息可以在各个监管机构( 可对保密性/ 可靠性负责) 之间,如国家局与省局、国家局与直属单位之间实现信息交互和数据共享,所提交的文件质量需保持一致。
2、CTD 与 eCTD的差异
CTD包含的5个模块中,除了第1个模块与eCTD的模块不一样,其他4个模块的信息是可以复用的。
CTD的第1个模块中,包括各地区特殊的文件,因此,提交的区域不同,要求也是不一样的。eCTD会根据所提交的不同要求,提供各种不同的模板提示。

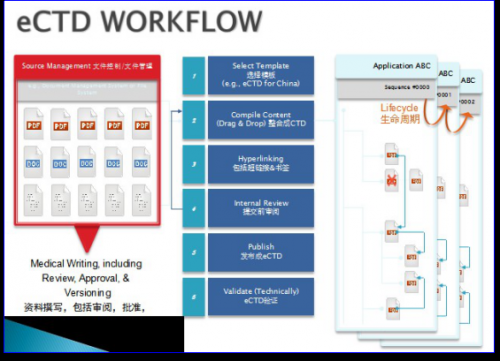
3、eCTD的定义
从IT的角度来讲,eCTD是以XML为骨架结构的PDF文件包,在NeeS的基础上加入了XML计算机语言,通过XML将大量的PDF文件有机结合在一起,并通过定义PDF文件的属性实现注册文件的生命周期管理。
三、IT人员应该做哪些准备
在监管越来越严格和统一的前提下,新药研发中的eCTD规范化将会越来越多的体现,药企的IT人员应该做好一些相应的准备。
数据层面:FDA在eCTD的很多标准、原则里面,提出了详细的数据标准。
软件层面:功能(如撰写模板、书签、超链接),更新升级,合规性,软件安装、验证及培训,多语言等。
网络层面:电子提交通道非常重要,其中的影响因素,包括电脑硬盘空间、数字证书、JAVA版本、浏览器版本、网络速度等。
企业系统架构规划:做CSV系统验证时,因为是标准的验证,不需要配置很多工作流。
软件维护:维保协议或自建软件维护团队,确保在递交的过程中遇到的技术问题能够得到快速响应。



